
The Biological and Environmental Research (BER) program supports scientific research and facilities to achieve a predictive understanding of complex biological, earth, and environmental systems with the aim of advancing the nation’s energy and infrastructure security. The program seeks to discover the underlying biology of plants and microbes as they respond to and modify their environments. This knowledge enables the reengineering of microbes and plants for energy and other applications. BER research also advances understanding of the dynamic processes needed to model the Earth system, including atmospheric, land masses, ocean, sea ice, and subsurface processes.
Over the last three decades, BER has transformed biological and Earth system science. We helped map the human genome and lay the foundation for modern biotechnology. We pioneered the initial research on atmospheric and ocean circulation that eventually led to climate and Earth system models. In the last decade, BER research has made considerable advances in biology underpinning the production of biofuels and bioproducts from renewable biomass, spearheaded progress in genome sequencing and genomic science, and strengthened the predictive capabilities of ecosystem and global scale models using the world’s fastest computers.
BER supports three DOE Office of Science user facilities, the Atmospheric Radiation Measurement (ARM) user facility, Environmental Molecular Sciences Laboratory (EMSL), and Joint Genome Institute (JGI). These facilities house unique world-class scientific instruments and capabilities that are available to the entire research community on a competitive, peer review basis. Additionally, four DOE Bioenergy Research Centers were established to pursue innovative early-stage research on bio-based products, clean energy, and next-generation bioenergy technologies.
BER Science Highlights
BER Program News
BER Subprograms
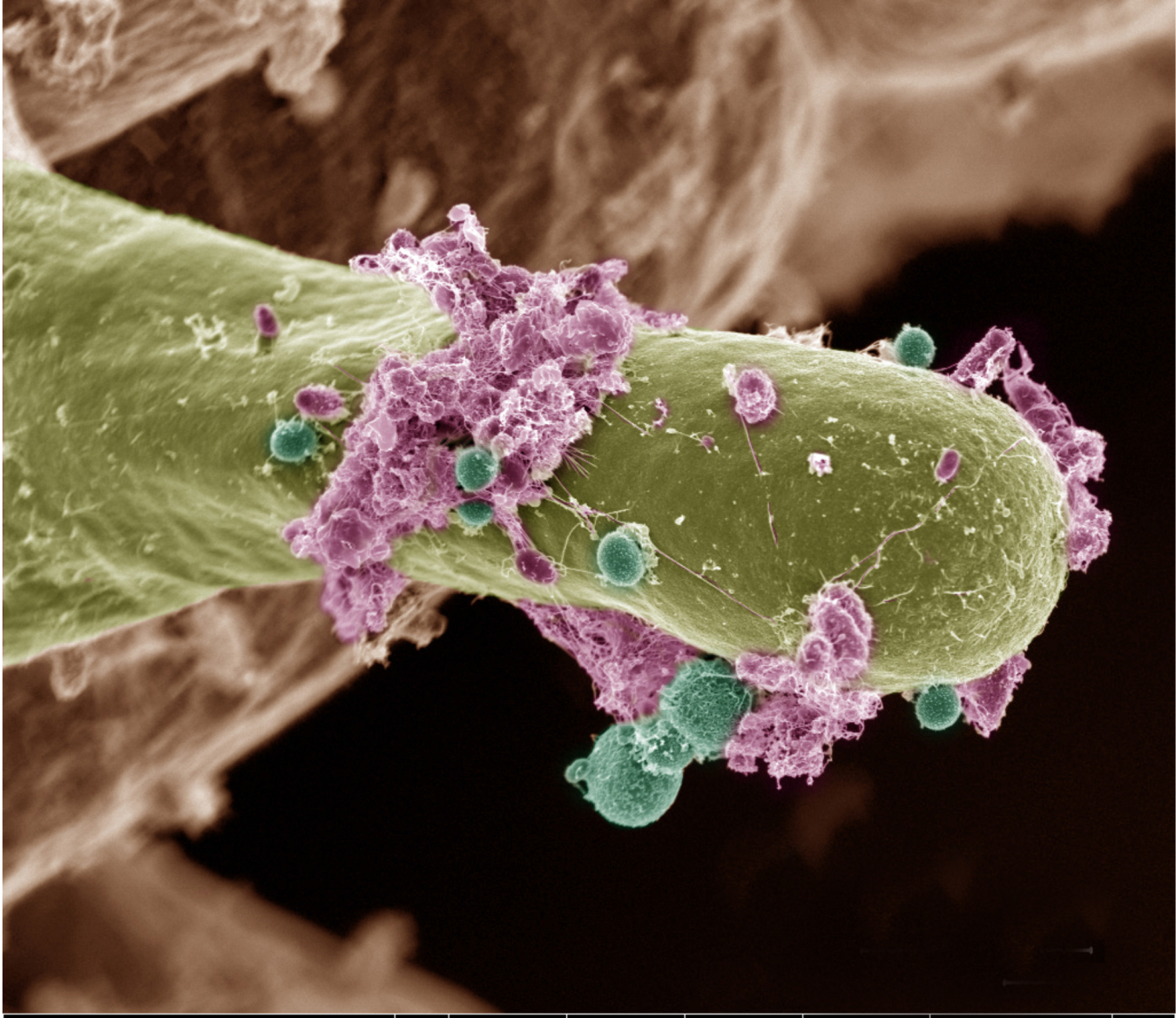
Biological Systems Science
Research to understand complex interactions that determine the function of biological systems, from single cells to plants.

Earth and Environmental System Sciences
Research on atmosphere, land, and water components and interactions that help inform regional and global earth system modeling.

DOE Bioenergy Research Centers
Supports fundamental research addressing the challenge of converting renewable plant biomass (non-food) to biofuels and bioproducts.
BER DOE Office of Science User Facilities

Atmospheric Radiation Measurement (ARM)
Observation network for understanding cloud and aerosol interactions with the Earth’s surface.

Environmental Molecular Sciences Laboratory (EMSL)
Houses more than 50 premier instruments and modeling resources that can be accessed to understand the physical, chemical, and cellular processes of biological and environmental systems.

Joint Genome Institute
Provides the research community with high throughput DNA sequencing, synthesis and analysis of plants, microorganisms and microbiomes in support of BER biological systems science research.
BER Research Resources



Contact Information
Biological and Environmental Research
U.S. Department of Energy
SC-23/Germantown Building
1000 Independence Avenue., SW
Washington, DC 20585
P: (301) 903 - 3251
F: (301) 903 - 5051
E: Email Us